The disease
DESCRIPTION
Primary ciliary dyskinesia (PCD), also called the immotile-cilia syndrome or Kartagener’s syndrome when associated with situs inversus) is characterized by chronic cough, chronic rhinitis, and chronic sinusitis due to abnormal function of the airways’ cilia (Fig. 1). Otitis, salpingitis, nasal polyposis are also common. The underlying cause is a defect of cilia in the airways, making them unable to beat (ciliary immotility), to beat normally (ciliary dyskinesia), or to be absent (ciliary aplasia). It is an inherited disease that has been described from most parts of the world occurring equally in men and women in approximately one in 10,000 to 30,000 individuals.
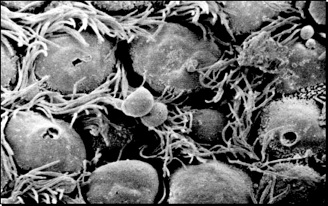
Figure 1 A: Scanning electron micrograph illustrating ciliary surface once the mucus has been removed. Bacteria, dust from the inhaled air, and dead cells from the deep lung are normally entrapped into the mucous and carried out to the larynx and mouth by the movement of the cilia (mucociliary escalator)
.png)
Figure 2 : Cilia float in the watery periciliary fluid and grip into and propel the superficial mucus blanket during the effective stroke. The mucus blanket is thus moved to the nasopharynx, from where it is swallowed together with adherent pollutants
Because the embryonic nodal cilia are also defective, migration of the thoracic and abdominal organs is random leading to inversed asymmetry (situs inversus totalis) in 50 percent of the individuals. When situs inversus, chronic sinusitis, and bronchiectasis occur together, an individual is said to have Kartagener's syndrome, a subgroup of primary ciliary dyskinesia occurring in around one in 20,000 to 40,000 individuals altogether. Bronchiectasis may develop in young persons but is never present at birth.
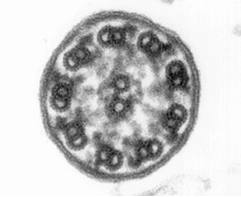
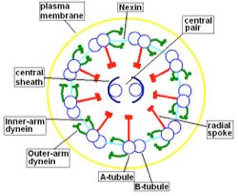
Morphological and physiological alterations — The fundamental defect in PCD is constituted by variable alterations in the axoneme, the core structure of airways’ cilia, spermatozoa’s flagella and embryonic nodal cilia. Airways’ cilia play a crucial role in mucous movement and clearance, the flagella are responsible for spermatozoa propulsion and embryonic nodal cilia contribute to the proper migration of the asymmetric organ (including heart, liver, stomach and spleen) during embryogenesis. The axoneme (Fig. 1) consists of a set of nine doublet microtubules - each of which consists of a partial microtubule connected to a complete microtubule - arranged cylindrically around a pair of singlet microtubules. These microtubules are arranged with their plus ends at the tip of the axoneme and their minus ends anchored in basal bodies in the cell. The axonemal dynein motors are distributed along each doublet as inner and outer rows of arms. PCD is a highly heterogeneous syndrome because it can be caused by a defect in any of the many proteins constituting the cilia (or sperm flagella), or in those needed for the proper assembly of cilia. Different protein components (including in particular dynein protein) may be altered or missing in different patients, and different clinical manifestations may develop depending upon the exact nature of the lesion. Conversely, different families may have protein alterations (different mutated genes) even though clinical symptoms are identical.
PATTERN OF INHERITANCE
Primary ciliary dyskinesia is inherited as an autosomal (modified genes on non sexual - X or Y - chromosomes) and recessive (modified genes on chromosomes from both parents are needed) disease.
Given that many structural and functional proteins of the axoneme can be altered, a high number of different mutations on different genes is suspected. In only a few cases, has it been possible to identify a specific chromosomal locus and gene product. As an example, a locus on chromosome 5p encoding an axonemal dynein heavy chain protein called DNAH5 was isolated on the basis of linkage analysis from 25 separate families. From this group, four homozygous and six heterozygous mutations of DNAH5 were identified, all in association with clinical primary ciliary dyskinesia and ultrastructurally abnormal outer dynein arms. Complete absence of DNAH5 along the ciliary axoneme results in immobility, while absence of DNAH5 in the distal portion of the axoneme causes impaired mobility.
The occurrence of situs inversus apparently has an element of random determination. Rather than having one gene for situs solitus (organs in their normal position) and one for situs inversus, the nodal cilia of the embryo are responsible for controlling the normal position of heart and visceral organs, and without such control there is an equal chance of situs inversus and situs solitus. Two pairs of identical twins with primary ciliary dyskinesia have been identified with one twin presenting situs inversus and the other one situs solitus.
CLINICAL MANIFESTATIONS
There is considerable variation in the clinical presentation of primary ciliary dyskinesia, although the most common features are recurrent respiratory infections otitis and sinusitis.
Pulmonary findings
Newborns with primary ciliary dyskinesia often suffer from mild respiratory distress, and a constantly runny nose may be noted beginning in early childhood. An increased incidence of respiratory infections with chronic cough and expectoration of sputum, often purulent, is commonly encountered as the infant grows. These symptoms tend to increase during the course of the day rather than peak in the morning, as they do in smoker's bronchitis. Nasal polyposis is frequently present. In contrast, uncomplicated common colds do not seem to occur more often than in normal subjects, nor do they usually have a more severe course.
Cylindrical or saccular bronchiectasis may occur, even in childhood, and usually affects the middle and lower lobes of the lungs. Patients with bronchiectasis generally manifest auscultatory crackles and may have wheezes which mimic asthma, particularly in children.
Common findings on chest radiograph and CT scan are a moderate degree of hyperinflation, peribronchial thickening, atelectasis, and bronchiectasis. Bronchiectasis involves the lower lobes almost exclusively and has been reported to occur in approximately 50 percent of patients [14]. Multiple, diffuse small centrilobular nodules up to 2 mm are sometimes seen, probably representing bronchiolitis [15].
Spirometry often reveals mild to moderate airway obstruction with variable responsiveness to bronchodilators. The major infecting bacteria are Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, and less commonly, Pseudomonas aeruginosa.
Sinuses
Sinusitis is present in the maxillary, ethmoidal, and frontal sinuses, although the frontal sinuses often fail to develop. The absence of a frontal sinus often gives the voice a somewhat nasal tone. Chronic secretory otitis media with recurrent acute exacerbations is constantly present during childhood and adolescence, but these problems become much less frequent following puberty. Sequelae of conductive hearing loss are common.
Situs inversus
Situs inversus, when present in patients with primary ciliary dyskinesia, occurs normally as a complete reversal of the circulatory system and the viscera. This has no serious adverse health consequences per se, and the condition often goes undetected until a chest radiograph is obtained. Situs inversus is a very useful sign when primary ciliary dyskinesia is being considered, but as described above, is present only in approximately 50 percent of patients with primary ciliary dyskinesia. Isolated situs inversus has a prevalence of about one in 10,000 in Scandinavia, and only about 20 to 25 percent of persons with situs inversus have chronic bronchitis and sinusitis [16].
Central nervous system
Fatigue and headaches are common complaints and may be caused by chronic sinusitis, although the headaches may persist even during infection-free periods. Hydrocephalus has been described from several persons with primary ciliary dyskinesia, and impaired function of ependymal cilia may be at least partially responsible [17-19].
Fertility
Most men with this syndrome have living but immotile spermatozoa and are infertile, although some have motile spermatozoa but immotile cilia [20]. Women likewise have decreased fertility, with fewer than 50 percent successfully completing pregnancy [2,21].
Associated abnormalities
A number of other congenital abnormalities are occasionally associated with primary ciliary dyskinesia, including transposition of the great vessels and other cardiac abnormalities, pyloric stenosis, and epispadias.
DIAGNOSIS
A number of diagnostic tests have been developed to demonstrate the immotile, dysmotile, or missing cilia.
Measures of mucociliary clearance
It is logical to examine ciliary motility in vitro or to measure the mucociliary transport rate in the airways, but there are difficulties with both methods. Cilia from the airways may be immobilized by bacterial toxins; immotility is then acquired rather than inborn and is associated with a different prognosis. Furthermore, mucociliary transport involves a two-component system, and the abnormality may reside in the mucus rather than in the cilia. As an example, patients with cystic fibrosis generate very viscous mucus, which their normal cilia are unable to propel forward. Mucus from patients with asthma also is more viscous than in healthy persons.
Mucociliary transport can be measured in situ by administering an aerosol of small Teflon particles tagged with the short-lived isotope 99Tc [22]. Radioactivity within the lungs is then measured repeatedly for 2 hours by profile scanning of the thorax while the patient is supine. The patient must not cough during these 2 hours, because coughing acts as a substitute for mucociliary clearance and results in elimination of the isotope from the lungs. An absence of mucociliary clearance is a sign of ciliary immotility, dysmotility, or aplasia, which may be inborn or acquired.
A somewhat simpler and more rapid technique consists of depositing small grains of saccharin particles (or dye particles) in the inferior concha and measuring the time required for the taste of the marker to be perceived or for the dye to become visible in the throat. This technique appears less reliable than other methods of diagnosis and cannot be used in small children [23]. Alternatively, tagged albumin droplets are deposited in the nasal passages, and the rate of transport of the radioactive area is measured externally [24,25]. The sensitivity of the test is high but its specificity is low; therefore, a normal test result excludes PCD.
Assays for cellular rotation
A different method in which the rotation of nasal epithelial cells is assessed seems to circumvent some of these difficulties [26]. With this technique, epithelia from the inferior concha of the nose are taken with a biopsy brush and maintained in tissue culture for several weeks. The culture medium is then supplemented with pronase (to separate the individual cells from the epithelium) and with antibiotics (to eliminate any epithelial bacteria). After several days, cilia are shed and new ones emerge. Healthy cilia are then recognized by their ability to rotate the cells in the tissue medium, whereas the cells will not rotate if primary ciliary dyskinesia is present.
Biopsies
Biopsies can be obtained endoscopically from the bronchi or nasopharynx, or taken with a curette or preferably with a brush from the inferior concha of the nose. The biopsy should be rapidly transferred to an isotonic saline solution and examined in the living state, preferably using a video recording system to determine whether cilia have normal coordination, beat frequency, and beat pattern. However, such an investigation cannot distinguish between primary and secondary ciliary dyskinesia.
Alternatively, the fresh biopsy can be immersed in a glutaraldehyde solution and further processed for ultrastructural investigation. Cross-sections of the cilia are examined by electron microscopy, and specific defects or normal ultrastructure can be visualized (show figure 1A-1C) [27]. The diversity in ciliary ultrastructure among different patients with the same clinical symptoms is due to the fact that the immotile-cilia syndrome is a heterogeneous disease, and the underlying defect may reside in any of a great number of ciliary genes.
A commonly observed defect is the absence of so-called dynein arms (dyneins are the high molecular weight motor proteins responsible for ciliary motility); other reported ultrastructural abnormalities include the absence of radial spokes, absent, or additional microtubule assemblies. A variant form of ciliary transposition, which results in circular ciliary beating and central microtubule agenesis, has also been described [28].
In 10 to 20 percent of cases, the living cilia are seen to be immotile but the ciliary ultrastructure appears quite normal; this likely reflects an underlying defect in membrane pumps or in other proteins not visible in electron micrographs. Use of computer-based image processing algorithms can improve visualization of ultrastructural abnormalities detected using electron microscopy [29].
Other tests
Other diagnostic tests have been proposed but have a less well-defined role. These include:
-
- A gel electrophoretic examination of dyneins in a biopsy from the nasal epithelium or a sample of ejaculate [30].
- Using a dynein gene probe to map the corresponding messenger RNA in lung or testicular tissue [31].
- Measuring the amount of exhaled nitric oxide, which is very low or absent in patients with PCD but high in certain other lung diseases, such as asthma and bronchiectasis [5,32-35]. (See "Exhaled nitric oxide analysis").
- Examining whether spermatozoa are motile or immotile, although care must be taken to differentiate dead from living but immotile spermatozoa. It must also be kept in mind that occasional patients have immotile cilia but normally motile spermatozoa [20].
References
1. Afzelius, BA. A human syndrome caused by immotile cilia. Science 1976; 193:317.
2. Afzelius, BA, Mossberg, B, Bergström, SE. Immotile-cilia syndrome (primary ciliary dyskinesia) including Kartagener syndrome. In: Scriver, CR, Beaudet, AL, Sly, WS, Vale, D (Eds), The Metabolic and Molecular Bases of Inherited Disease, 8th ed, McGraw Hill, New York, 2000.
3. Afzelius, BA, Stenram, U. Prevalence and genetics of immotile-cilia syndrome and left-handedness. Int J Dev Biol 2006; 50:2132.
4. Nonaka, S, Shiratori, H, Saijoh, Y, Hamada, H. Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature 2002; 418:96.
5. Noone, PG, Leigh, MW, Sannuti, A, et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med 2004; 169:459.
6. Sturgess, JM, Thomson, MW, Czegledy-Nagy, E, Turner, JAP. Genetic aspects of immotile cilia syndrome. Am J Med Genet 1986; 25:149.
7. Rosenbaum, JL, Cole, DG, Diener, DR. Intraflagellar transport: The eyes have it. J Cell Biol 1999; 144:385.
8. Pennarun, G, Escudier, E, Chapelin, C, et al. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am J Hum Genet 1999; 65:1508.
9. Olbrich, H, Haffner, K, Kispert, A, Volkel, A. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left--right asymmetry. Nat Genet 2002; 30:143.
10. Fliegauf, M, Olbrich, H, Horvath, J, et al. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am J Respir Crit Care Med 2005; 171:1343.
11. Noone, PG, Bali, D, Carson, JL, et al. Discordant organ laterality in monozygotic twins with primary ciliary dyskinesia. Am J Med Genet 1999; 82:155.
12. Bush, A, Cole, P, Hariri, M, et al. Primary ciliary dyskinesia: Diagnosis and standards of care. Eur Respir J 1998; 12:982.
13. Chapelin, C, Coste, A, Millepied, M-C, et al. Incidence of primary ciliary dyskinesia in children with recurrent respiratory diseases. Ann Otol Rhinol Laryngol 1997; 106:854.
14. Nadel, HR, Stringer, DA, Levison, H, et al. The immotile cilia syndrome: Radiological manifestations. Radiology 1985; 154:651
15. Homma, S, Kawabata, M, Kishi, K, et al. Bronchiolitis in Kartagener's syndrome. Eur Respir J 1999; 14:1332.
16. Torgersen, J. Transposition of viscera, bronchiectasis and nasal polyps. Acta Radiol 1947; 28:17.
17. Greenstone, MA, Jones, RWA, Dewar, A, et al. Hydrocephalus and primary ciliary dyskinesia. Arch Dis Child 1984; 59:481.
18. Picco, P, Leveratto, L, Cama, A, et al. Immotile cilia syndrome associated with hydrocephalus and precocious puberty: a case report. Eur J Pediatr Surg 1993; 3 Suppl 1:20.
19. De Santi, MM, Magni, A, Valletta, EA, et al. Hydrocephalus, bronchiectasis, and ciliary aplasia. Arch Dis Child 1990; 65:543.
20. Jonsson, MS, McCormick, JR, Gillies, CG, Gondos, B. Kartagener's syndrome with motile spermatozoa. N Engl J Med 1982; 307:1131.
21. Greenstone, M, Rutman, A, Dewar, A, et al. Primary ciliary dyskinesia-cytological and clinical features. Q J Med 1988; 67:405.
22. Camner, P, Mossberg, B, Afzelius, BA. Measurements of tracheobronchial clearance in patients with immotile-cilia syndrome and its value in differential diagnosis. Eur J Respir Dis Suppl 1983; 127:57.
23. Karja, J, Nuutinen, J. Immotile cilia syndrome in children. Int J Pediatr Otorhinolaryngol 1983; 5:275.
24. De Boeck, K, Proesmans, M, Mortelmans, L, et al. Mucociliary transport using 99mTc-albumin colloid: a reliable screening test for primary ciliary dyskinesia. Thorax 2005; 60:414.
25. Andersen, I, Proctor, DF. Measurement of nasal mucociliary clearance. Eur J Respir Dis Suppl 1983; 127:37.
26. Jorissen, M, Willems, T, Van der, Schueren B, et al. Ultrastructural expression of primary ciliary dyskinesia after ciliogenesis in culture. Acta Otorhinolaryngol Belg 2000; 54:343.
27. Afzelius, BA. The immotile-cilia syndrome: a microtubule-associated defect. CRC Crit Rev Biochem 1985; 19:63
28. Stannard, W, Rutman, A, Wallis, C, O'Callaghan, C. Central Microtubular Agenesis Causing Primary Ciliary Dyskinesia. Am J Respir Crit Care Med 2004; 169:634.
29. Escudier, E, Couprie, M, Duriez, B, et al. Computer-assisted analysis helps detect inner dynein arm abnormalities. Am J Respir Crit Care Med 2002; 166:1257.
30. Mencarelli, C, Tiezzi, A, Ruggiero, P, et al. Heterogeneous localization of epitopes along axonemes of mammalian cilia. Biol Cell 1995; 83:179.
31. Kastury, K, Taylor, WE, Shen, R, et al. Complementary deoxyribonucleic acid cloning and characterization of a putative human axonemal dynein light chain gene. J Clin Endocrinol Metab 1997; 82:3047.
32. Corbelli, R, Bringolf-Isler, B, Amacher, A, et al. Nasal nitric oxide measurements to screen children for primary ciliary dyskinesia. Chest 2004; 126:1054.
33. Grasemann, H, Gärtig, SS, Wiesemann, HG, et al. Effect of L-arginine infusion on airway NO in cystic fibrosis and primary ciliary dyskinesia syndrome. Eur Respir J 1999; 13:114.
34. Horvath, I, Loukides, S, Wodehouse, T, et al. Comparison of exhaled and nasal nitric oxide and exhaled carbon monoxide levels in bronchiectatic patients with and without primary ciliary dyskinesia. Thorax 2003; 58:68.
35. Csoma, Z, Bush, A, Wilson, NM, et al. Nitric oxide metabolites are not reduced in exhaled breath condensate of patients with primary ciliary dyskinesia. Chest 2003; 124:633.